EXtended Electron Distribution. (XEDs).
Since reliable quantum calculations are too slow to construct on many conformations, we have developed an alternative way of describing electron charge distribution which has been shown to reproduce a) intermolecular interations in accordance with experiment and b) electrostatic extrema generated by quantum mechanical DMA methods. These extended electron distribution points (or XEDs) are added to the target molecule before field are added.
A typical arrangement is shown for an aromatic system and a carbonyl group. 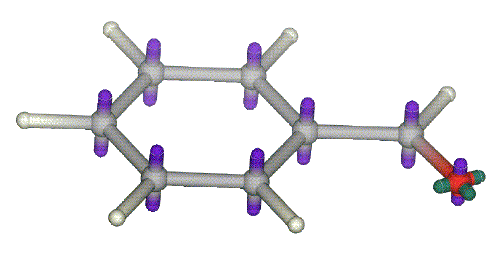 All the usual atoms to be found in organic chemistry have been implemented and the methodology has been validated using experimental data from a variety of intermolecular non-bonding interactions (J G Vinter, (1994), 'Extended electron distributions applied to the molecular mechanics of intermolecular interactions', J Comp-Aid Mol Design, 8, 653-668.). Atom-centred charges do not produce good field points and XEDs are recommended.
All the usual atoms to be found in organic chemistry have been implemented and the methodology has been validated using experimental data from a variety of intermolecular non-bonding interactions (J G Vinter, (1994), 'Extended electron distributions applied to the molecular mechanics of intermolecular interactions', J Comp-Aid Mol Design, 8, 653-668.). Atom-centred charges do not produce good field points and XEDs are recommended.
There is no provision in the XED molecular mechanics regime for any out-of-plane bending correction. Without the addition of XEDs, systems like the carbonyl group or the peptide link, will be minimised out of plane. Their observed planarity is believed to be the result of the overlap of pi-type orbitals. The addition of XEDs to the molecular mechanics formalism is an attempt to simulate these 'real life' orbitals. Further intramolecular minimisation with XEDs corrects any bending that may have occurred.
When you click on 'Add/remove XEDs', the system will not encourage you to proceed without having added partial atom centred charges (ACCs) beforehand. Add these with the 'Minimise/Charge' button and proceed to add XEDs after returning without further minimisation. When you re-enter the minimiser, it will know that you have added XEDs and will move directly to the minimisation step. See Appendix 4 for details of the minimiser and the limitation of the procedure using XEDs.
XEDs are not added to Carbon, Hydrogen or any atom with an electronegativity less than carbon. Halogens and sulphur are still the subjects of research (July 1996)
XEDs were designed to allow more accurate electrostatic field calculations without resorting to wave functions. It is therefore important that XEDs are added before you calculate these fields points. (See Section 2 for details about Field Calculation and Usage.
APPENDIX 4 XED/COSMIC
MINIMISATION
It is accepted that the limitations of molecular mechanics are inherent in the concepts. Any degree of accuracy can be incorporated if enough terms are added and the potential regime limited to a select group of highly related molecules. This is so for any empirical predictor. In essence, the molecular mechanics method is flawed and as such, should be used with caution and yet approached with a sense of humour. It cannot ever be a serious answer to molecular structure.
Fifteen years ago, the original COSMIC force field was designed to handle a broad spectrum of small molecules (mw 50 - 1000) expected to be found in a medicinal chemistry environment. The same criterion hold today. COSMIC uses the minimum number of essential potentials, the smallest possible set of atom types, the simplest of force field parameters and the least number of 'fudge factors' as is commensurate with reasonable accuracy. The bond and angle energy terms are both a 'Hooke's Law' potential. The torsional parameter is the first term only of a Fourier series, the van der Waals energy is calculated using a Morse potential and the coulombic energy is the standard charge product divided by distance and the 'dielectric'. There are no specific terms for out-of-plane bending or hydrogen bonding. There are no special arrangements for the strain in non-six-membered rings, no bond or angle cross terms and a simple two-atom definition for torsional bonds. The internal Huckel calculation ensures that the system gets some idea of bond order and this is used to adjust bond and torsional parameters. In this way, force field entries can be simple but geometries will incorporate the differences between the endo- and exo- aromatic bond, the degree of conjugation in a double or amide bond and so on. XEDs, which have no vdW component and affect only the coulombic term, are optional additions which adjust out-of-plane bending, refine electron distribution for better coulombic interactions and improve recognition in complex formation.
The COSMIC force field has behaved very well over the years and has usually kept faith with intuition. Testing a force field is always a tricky problem because the experimental data are often special cases, subject to solvent effects, crystal forces, contaminants and other influences. All we can do is to stick to the Modellers' Commandments: 'Compare like with like. Study relative relationships. Do not mix methodology within a project. Be wary of your own prejudices and those of others'.
- The detailed workings of the energy calculation are reported in:
- S D Morley, R J Abraham, I S Haworth, D E Jackson, M R Saunders, and J G Vinter, (1991), 'COSMIC90- an improved molecular mechanics treatment of hydrocarbons and conjugated systems', J Comp-Aid Mol Design, 5, 475-504, and references therein.
- J G Vinter, (1994), 'Extended electron distributions applied to the molecular mechanics of intermolecular interactions', J Comp-Aid Mol Design, 8, 653-668.
This is the minimiser regime invoked when you click on the 'minimise/charge' button in Book 8 of the main graphical screen. By default, it is a conjugate gradient procedure with added torsional control. The energy of a structure (as calculated by the sum of 5 energy potentials using the force field databases held in the directory FFF), is minimised with respect to the Cartesian coordinates of each of its atoms. The force field has been described in the literature cited above.
Although modern minimisation techniques such as SIMPLEX or Conjugate Gradient are very good at reducing a structure to an acceptable local minimum geometry, it is quite difficult to get a minimiser to twist a torsional angle in an attempt to refine its answer a little. The XED minimiser has been modified so that after it has reached its limiting energy by atom movement, it switched to torsional twisting minimisation to see if it can do better. If the energy keeps dropping, it will switch again to atom relaxation to refine the new torsional setting. The alternation between torsional twist and atom relaxation will continue until the energy reaches a plateau and the system prompts you for 'more minimisations'. It is often wise to say 'Yes' at this point and do another iteration round. Continue to say 'Yes' until no more iterations are forthcoming. The reason for this odd ability for the minimiser to carry on just when it thought it had finished, is probably to do with rounding errors in data transfer and the re-establishment of initiator values at the start of minimisation.
You may ask, why not put XEDs on to begin with? Why bother with an ACC minimisation step. It transpires that the addition of XEDs (more points!) greatly increases the complexity of the conformation hypersurface. In other words, the number of local minima available to the molecule soars. As a consequence, XED minimisations tend to finish too early and get trapped in some high energy local minimum.
It is worth remembering that the mathematical minimiser will only be able to give you the best local minimum geometry which is closest to the structure it started with, rather like Transition State Theory. Unless you are very lucky, the result will not be a Global Minimum- and how would you know what the Global Minimum looked like anyway? - certainly not definitively from X-ray data. The importance of the Global Minimum of a drug or hormone has receded in recent years due to the revelation that it is rarely found in the active sites of receptors and enzymes.
The only way to overcome the 'local minimum' trap is to undertake a conformation hunt. Even this procedure will not guarantee a comprehensive list of all possible conformers nor a Global Minimum nor yet any idea of the ease of interconversion of conformers. However, it is better than relying on the 'One Shot' which should be retained for building purposes only.
Since exhaustive conformation hunting is not possible for the majority of interesting molecules because the combinatorial problem is so large, most realistic hunters use approximate stochastic approaches. In general, the problems to be addressed by a conformational hunter are: to cover as much conformational space as possible, to handle torsional variations in flexible chains, know about conjugation and deal with ring flexibility. The limitations that the hunter must be put under in order to produce a useful answer in a reasonable time are: the energy required (do you want THE global minimum), the length of time allowed to look over conformational space, the precision of the torsional twist increment, how many conformers to keep and within what energy range should the conformers be collected.
The XED conformational hunter starts from a single arbitrarily minimised structure with ACCs. If flexible rings are present, it is first subjected to a chosen number of torsional dynamics cycles, each over 1000 cycles with a structure collection at 20 cycles t a temperature of 10,000K. Each collected structure is minimised by CG (see single Shot Minimisation in b) above) and held in a 'list' which has been filtered for duplicates (least squares coordinate rms fit < 0.25). This procedure hopefully picks up all ring local minima. If there are no flexible rings, no dynamics needs to be done and this 'list' contains the input starting structure alone.
The 'list' is used to start a 'torsional' hunt. Every flexible bond (and non-flexible bond if you choose) is turned through 360 degrees by a user-defined step and all minima found during the twist are stored. This process is iterated to 0.01kcal/mole. The molecule is torsionally randomised and the process is repeated. The number of torsional randomisations is also chosen by you. The number of stored conformers must fall within an energy range (defined by you) above the current 'global minimum'. Eventually, this number reaches a ceiling above which no more are held (2000 at present), even though the search continues. When the hunter has finished, the duplicate filter is applied and the resulting conformers are stored in a .ast file (i.e. a file extension of .ast).
The hunter then takes the .ast file and minimises all entries. After duplicate filtration, the minimised conformers are held in a .bst file. Next, the hunter adds XEDs to the .bst entries, minimises, filters and writes the unique XED structures into a .cst file.
Later, we will discuss the use of fields as similarity indices in drug design. The comparison procedures for this stage use .cst conformer collections after fields have been added to them. As a final stage, the hunter will put fields on all the entries in the .cst file if you ask it to. The final conformer/field file, containing representative conformers over a given energy range with XEDs and fields, is called the .fst file.
All these multi-files can be viewed in the main XED graphical screen

