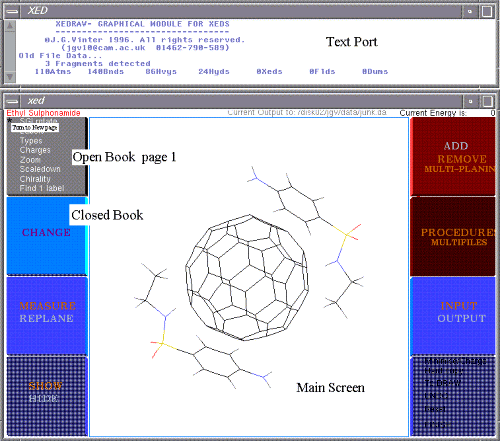
A small Text Port opens at the top of the screen and the main graphics window can be pulled out to suit your needs. Full-sizing is recommended, followed by small adjustments to both windows depending on your display unit. Both windows may be iconised if needed.
XED/COSMIC is continually being updated and there may be functions on screen that I have not had time to add to this document. Let me know if you encounter problems or omissions.
Books will be referred to by name or number:
Book 1 VIEW- Commands to view attributes and size display
Book 2 CHANGE - Commands to change molecular geometry or attributes
Book 3 MEASURE & REPLANE -
Book 4 SHOW & HIDE - All these commands are turned on and off via a toggled button.
Book 5 ADD & REMOVE - Add/Rem buttons are 'pseudo-toggled'; e.g. If a bond is there, it is removed. If not, a bond is added
Book 6 PROCEDURES -
Book 7 INPUT & OUTPUT -
Book 8 Open commands - This book is always open.
The best way to understand a package is to use it. Try the exercise in Appendix 1
If there is no current molecule in the running file, the system will display a default molecule.
Click the LEFT mouse button anywhere inside the display box (black area). The menu items on each coloured 'book' will disappear.
With LEFT mouse button (LMB) depressed- move cursor to rotate in x & y
With MIDDLE mouse button (MMB) depressed- move cursor to rotate in z
With RIGHT mouse button (RMB) depressed- move cursor to translate
Cursor movement is followed by the molecule.
To re-establish the Books and exit motion, move the cursor out of the display box. No click or button is necessary.
If there are more than one fragments in the display (see later Read (add a dat) in Book 7), a noise will be heard. Note the questions in the Text Port. You will be asked to LEFT-mouse-click on one atom of the fragment to be rotated. If you want all to rotate, LEFT-mouse-click on an empty part of the display.
