The Silicon Graphics Teaching Laboratory
has been replaced by the
Chemi
cal Information Laboratory
and this information is for historical interest only
MacroModel Notes
How to start MacroModel / How to stop MacroModel
When you have logged on, make a window, type rmmod. The computer will ask you
for a terminal type. Type 2, for '3D Silicon Graphics (GL)'. The program will now start, and
windows will appear on the screen, looking a little like this:
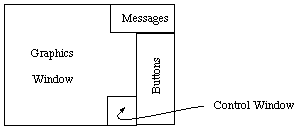
There are three windows: a large black one, which is the Graphics Window, a small
green/grey one in the top right of the screen, which is the Message Window, and a green/grey
one down the right hand edge of the screen, which is the Buttons Window.
Stopping MacroModel: Click on STOP in the lower left corner of the Buttons
window with the left mouse button, and confirm you really want to stop by clicking
Yes.
This stops MacroModel, but leaves you logged on to the Indigo. You logout from the Indigo
by choosing Log-Out from the Toolchest's System box.
General notes
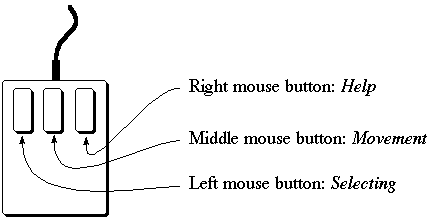
Use the left mouse button for picking atoms, bonds or points and pressing buttons.
The middle mouse button is used for rotating and moving molecules. The right mouse button
is used to provide help. If you click on any button with the right button, a new window will
appear explaining what the button does. Watch the message window; the program may want
to tell you something!
MacroModel has three modes: INPUT; ENRGY; ANLYZ.
Clicking on
these buttons will change the options available in the buttons window. INPUT is used for
drawing and editing molecules. ENRGY is used for calculating the energy of a conformation
of a molecule, and finding ways of altering the conformation to lower the energy.
ANLYZ is used to analyse the properties of the molecule, such as bond lengths,
bond
angles, etc.
How to draw a molecule
Choose the INPUT mode. Click on DRAW. Now click (left mouse button) in the
graphics window. You will draw a chain of carbon atoms. Hold the middle mouse button
down and move the mouse in the graphics window to rotate the chain. Some elements are
available in the buttons window. Choose an element and click on a carbon atom to change the
atom type.
It is also possible to draw molecules by using templates. Click on CH4, then click
in the Graphics window. A methane molecule should appear. Click on a hydrogen, and the
hydrogen turns into a saturated carbon, making an ethane molecule. There are many
templates available. If you click on PEPTID, NUCLEI, or CARBO
new templates
will appear.
You can mix these two methods of drawing molecules. Use the H ADD and H
DEL buttons to add and remove hydrogens from the structures. Try clicking twice on
these
buttons instead of once, to find out what happens. Click with the right mouse button to get
help.
Reading and Writing molecules
Once you have drawn a molecule, you want the computer to remember it. To do this,
you must WRITE it to a disk file. Click on WRITE and a dialogue box will
appear.
Enter a filename. Suitable names do not include spaces, or other punctuation, but can include
numbers, letters, hyphens, underscore '_'. You must be careful to have the mouse in the box
into which you are typing. When you have typed a name, click Write then
Quit to get
rid of the dialogue box.
To retrieve a molecule from a disk file, click READ. If you know the precise
name of the file, type it in and click on Read. Otherwise, click Open, and
you will be
shown a list of the available files.
Energy Minimisations
Once you have drawn a molecule, go into the ENRGY mode. Click on
Start to
begin an energy minimisation. You will be asked if you want to use an old or a new
command file. Choose New. You may be asked if the program should add lone pairs to the
structure. If this question appears, click Yes. You will then be asked for a filename. Any
alphanumeric string is acceptable. Capital letters are different from small letters. Do not have
a space, punctuation, or '*' as part of the name. If files already exist with the name that you
give, they will be overwritten.
The minimisation should now proceed. If there are any errors in the message window,
it may be because there are some hydrogens missing from the structure. Go pack into
INPUT mode, click twice on H Del, select All, click twice on H
Add, and select
All. Now try minimising again.
Pressing the left and right cursor keys start the molecule rotating.
Press the cursor keys a second time to stop this.
More Information
- MacroModel Atom Types
- New Batchmin System
- Files containing several structures are written in a new compressed
format. These can be converted into the old format by typing:
m54 <new format file> <old format file>
|