The Silicon Graphics Teaching Laboratory
has been replaced by the
Chemical Information Laboratory
and this information is for historical interest only
Practical One: Building Molecules
(1) Chair and boat cyclohexane
Start MacroModel running. Build a cyclohexane molecule in its chair
conformation (INPUT mode). Make sure all the correct hydrogen atoms
are present using H ADD and H DEL. Go into the
ENRGY mode and minimise the structure using the MM2 force field,
following the instructions at the end of this booklet (Remember to choose
New when asked if you want an Old or a New command file). Make a note
of the energy, then repeat the procedure for the boat conformation of
cyclohexane. Are the relative energies as you would expect?
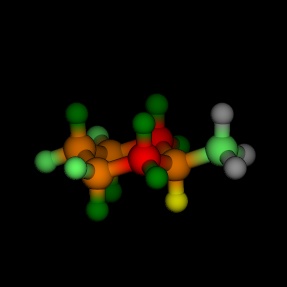
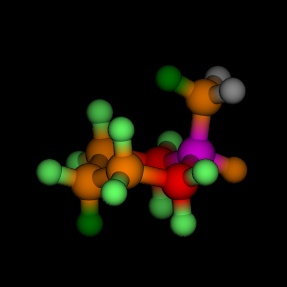
Equatorial and axial cyclohexane, coloured by the energy, as
calculated by MM2
(2) Measuring distances and angles
Go into the ANLYZ mode, and use ADist, BAngl,
DAngl to measure the bond lengths and angles of the two
molecules. How do they differ?
Click on Opt to view the molecule as a CPK model or a ball and
stick model. Is there a hole in the middle of cyclohexane?
(3) Equatorial and axial substituents on cyclohexane
Find the relative energy of equatorial and axial methylcyclohexane. Which
is the preferred conformation? How does this differ for phenylcyclohexane,
iso-propylcyclohexane and tertiary-butylcyclohexane? (Squillacote et al. J.
Am. Chem. Soc. 1975, 97, 3244)
(4) DNA manipulation
Build a DNA base pair by returning to INPUT mode, choosing the NCUCLEI
submode. Click on Ad then click somewhere on the screen (If you
click first on GROW and then choose the names of the bases, it
will grow a chain of DNA).
Go into the ENRGY mode. Try to minimise the base pair. You should first
get a message saying that the lone pairs mismatch. Ask the program to
correct this. Minimisation will start, but quickly stop with an error message.
This is because MM2 does not have the correct parameters for the
molecule. The main problem is that the molecule does not have all the
hydrogens drawn in. Some force fields, such as AMBER, can cope with
this, but MM2 cannot. You can add hydrogens by returning to the INPUT
mode, and clicking H Add three times. You should now be able to
minimise the molecule (New command file).
(5) Bredt's rule
Bredt's Rule states that double bonds will not form across bridgehead
carbons.
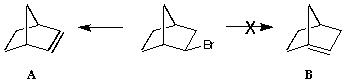
Build each of the unsaturated ring structures and record the energy of
each after full minimisation. Does this account for Bredt's rule?
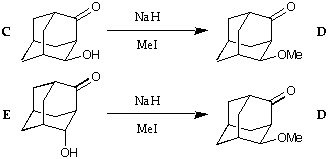
(6) An unusual rearrangement
The keto-alcohol C can be methylated to give the methylether D, as
expected. Surprisingly, the keto-alcohol E reacts to give the same product.
The rearrangement can be rationalised as a retro-aldol process preceding
the methylation. But what is the driving force for the rearrangement?
Calculate the energies of the products, or of suitable intermediates, to find
out.
(7) Protein structures
Click on READ. In the box that appears, click on PDB,
to read in Protein Databank Format files. Now Click on Open.
Move the mouse to the upper window of this new box, and type
/usr/local/brookhaven/*.ent. Click on Filter. A short list of
filenames should now appear. This is just a small selection of files in the
Brookhaven Protein Database.
Click on any of the names, then click Open Read.
The protein you have selected should appear on the screen.
Click on PEPTID, choose an amino acid from the list, then click
on any residue. This will perform site directed mutagenesis!
|