The Silicon Graphics Teaching Laboratory
has been replaced by the
Chemical Information Laboratory
and this information is for historical interest only
Practical Two: Conformation Searching
(1) Construct a Ramachandran type plot
MacroModel can construct Ramachandran plots for molecules. This
requires a large number of calculations, so it is best to choose a simple
molecule, such as one of those in the previous exercise. Start with a
minimised structure, go to the ENRGY mode, and click on Drive.
You will be asked how many angles you wish to rotate. Choose '2'.
The program will ask you to define two torsion angles, by clicking on four
atoms to define each angle. It will also ask for the rotational increment.
The default is 30o, but you can save a lot of time by entering 60o instead.
Now click on Start to initiate the calculation. The computer will
now calculate the molecule's energy for all points on the grid. If you chose
60o resolution for both angles, this requires thirty-six energy minimisations.
When the calculation is complete, go to the ANLYZ mode and click on
Plt2D (in versions of MacroModel before V4.5, this is the Cntr (contour) button). A new window will appear. Click on Open
to open the file containing the results of the previous calculation. The file
will be called filename.grd. Select this and click on Process. This
will draw a colour-coded Ramachandran plot.
Clicking anywhere on the plot with the middle mouse button will show you
the structure that corresponds to that point.
(2) Cram's rule
Non-chelating nucleophilic attack on these two diastereomeric aldehydes
(1, 2) gives opposite selectivity, even though Cram's rule states that the
selectivity should just depend on the a-centre. MacroModel is used to
suggest an explanation for this, based on the MM2 force field.
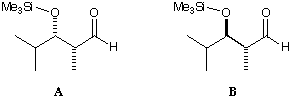 Draw the two molecules in MacroModel's DRAW mode. WRITE them to
the files: syn_aldehyde and anti_aldehyde. Minimise each molecule using
the MM2 force field. This will give you local minima.
Draw the two molecules in MacroModel's DRAW mode. WRITE them to
the files: syn_aldehyde and anti_aldehyde. Minimise each molecule using
the MM2 force field. This will give you local minima.
To discover the preferred conformation of these molecules (the global
minimum and any low energy conformations), a conformational search is
necessary. Check you are in the ENRGY mode, select the CSRCH sub-
mode (this is for Conformational Searching), and click on MCRLO. This
will set up a Monte-Carlo conformational search (Described in J. Am.
Chem. Soc. 1990, 112, 1419 - 1427). You will be given various options.
Choose 'Automatic setup.' This will set up a Monte-Carlo search, by
deciding which torsion angles need to be varied to investigate the
conformational space of the molecule. You will be presented with two
more dialogue boxes. Click OK in both of them. Selecting
TBond will show you which bonds are being rotated (Has the
program chosen well?). Click Check to provide a summary of the
search and to check there are no mistakes.
Return to the minimise (MINIM) sub-mode, and click START to set the
conformational search going You will have to give the search a name, for
example: anti_aldehyde_mc. The search will take some time. The search
will create a number of files, including: anti_aldehyde_mc.dat,
anti_aldehyde_mc.com, anti_aldehyde_mc.out, anti_aldehyde_mc.log.
The file which ends .log can tell you how the conformational search is
progressing. The job will continue if you do something else with
MacroModel, and even if you log out (New command file).
If you have stopped MacroModel running, try cat anti_aldehyde_mc.log or
more anti_aldehyde_mc.log to see how things are going. If you have
MacroModel running, go into the ENERGY mode, and click on MONTR, to
monitor how the search is progressing. The search will find the global
minimum structure more quickly if it began with a low energy structure.
Usually it is not a good idea to ask the computer to do more than one
thing at a time, because it spends too much of its time swapping between
jobs, and not enough time working. The computer can just about cope with
two things at a time, so for this practical you can start the syn and the anti
conformation searches together.
You are trying to find the Global minimum energy for each structure.
When the search is finished (one thousand structures investigated), you
should have found the lowest energy structures several times each. This
indicates that you have probably found the global minimum. If you have
only found the lowest energy structure once, the search may not be
complete. This would probably be because the search began with a very
strained structure. If this happens, restart the search, beginning with the
lowest energy structure you have.
When the search is complete, you should have a list of conformations and
their energies. You can examine the results by READing in the structures
from the anti_aldehyde_mc.out file. The energy of each structure will
appear in the message window as you read in the structure.
Examine the low energy structures of 1 and 2. Which face of each
aldehyde is more open to attack?
The conformation searches are unlikely to be completed in the time
allotted to the practical. However, they will probably progress far enough
to suggest an explanation for the surprising selectivity of nucleophilic
attack. Leave the searches running at the end of the practical. They will
probably be complete by the next session.
Questions (a) what evidence do you have that you found the global
minimum structure? (b) predict the stereochemistry of the product of
nucleophilic attack on each of the aldehydes (c) explain why Cram's rule
may not apply to both these diastereomers
(3) Erythromycin seco-acid
The results of a conformational search on an analogue of the seco acid of
erythromycin are in the file /usr/local/examples/erythro_mc.out and
/usr/local/examples/erythro_h2o_mc.out.The former was done in vacuo
and the latter was done using additional equations which allow for the
effect of water on the molecule. How do the results differ?
This is the global minimum structure for the
conformation search without a solvent model. The structure changes
dramatically, when a solvent model is applied.
|