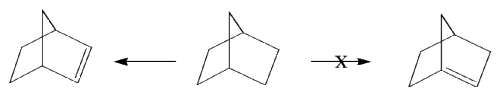
This exercise is to demonstrate how the DRAW window works and tests whether the modelling system can verify Bredt's Rule
Bredt's rule states that a double bond will not form across a bridgeheadcarbon.
Structure 1.......................................................................................Structure 2
Enter DRAW by picking 'To DRAW'. 'Clear' the screen. 'Yes' you do'
Create an atom on the blank screen using the right mouse button (RMB).
Create the next atom 2 inches away using the RMB. Atom 1 and atom 2 are bonded.
Continue creating the norbornene in this way. (Any shape will do so long as the bonding is correct). You will need to start a new node that is not bonded to the last atom you created. Pick 'POINT' with the RMB then place the atom or click on an existing atom.
If you create an unwanted atom, click the 'Remove 1 atom' button with the LMB and click on the unwanted atom with the LMB. If you create an unwanted bond, use 'Remove 1 bond' and click on the two atoms making up the bond in the same way. If you want to start again, 'Clear' the screen. For fun, click on an atom with the MMB and pull it about. You can improve the look of the 2D drawing this way and untangle confusions.
Click on Csp2 in the left hand long book with the LMB. It will disappear.
Click on one of the atoms making up the double bond with the LMB and the Csp2 will appear at that atom. Repeat for the other double bond atom.
The molecule is complete- but not chemistry! Rotate the drawing by double-clicking in the display area with the LMB and moving the cursor with the LMB depressed. Return to the main window by moving the cursor outside the display area with the LMB released
Click 'To View'
Pick the 'Minimise/Charge' button with the LMB in the open Book 8 and reply 'N' (or default i.e. hit <Enter>) to the question 'Add partial charges''. A new question appears asking if you want to minimise some or all the atoms. Reply 'Y' (or default) to this question.. A list of the bonds, angles, torsions and vdWs are determined and the Force Field parameters are retrieved. Reply 'N' (or default) on the next question about turning off torsional minimisation. You will be asked about the Dielectric Constant, the Accuracy and how many iterations you want, - Default on all by pressing <Enter> at each prompt. Watch the Energy and RMS drop. Questions at the end of the minimisation can be defaulted (hit <Enter>).
Add hydrogens by placing the cursor over Book 5 and activating page 1 item 1 - the 'Add/Remove H' button and reminimise using the 'Minimise/Charge' button in the open Book 8.
This time, when you are asked whether you want to CHARGE the molecule enter 'Y' (it is not usually necessary to do so with such a non-polar molecule).
Default on all other questions.
The minimiser should terminate before 200 iterations Note down the ENERGY [18kcals/mole or so?] look at the various energy contributions from bond angle, vdW etc. and return to the display box by defaulting on any other questions
Save the molecule using WRITE in Book 7. Enter ' xx_BRED1' in reply to 'Filename:', where xx are your initials.
Repeat the process to create the other structure.
(You can be clever about this by using the structure on screen (or reading in xx_BRED1) and modifying it. Hints: Remove hydrogens, move into DRAW, reallocate Csp2 and Csp3 atoms. Return to View and add hydrogens. You need to minimise again of course and note the energy [55kcals/mole or so?])
At the end of the exercise, you will have stored two files called xx_BRED1.DAT and xx_BRED2.DAT (.DAT is implied on both READ and WRITE)
You will have an energy value for each. Which is less stable, the bridge-head double bond or the non-bridgehead double bond? How do the dipole moments compare? Is there any distortion on the bridgehead molecule? What did you learn from looking at the individual energy contributions listed in the Text Port after each minimisation?
Sample of Approximate Energies (kcal/mole) after Minimisation of Structures 1 and 2
Structure | Coulombic | vdW | Bond | Angle | Tors | TOTAL |
1 | -3 | 2 | 0 | 17 | 3 | 19 |
2 | 3 | 3 | 0 | 20 | 32 | 52 |
