The inclusion of this module is essentially for training purposes. XED/COSMIC does not use QM in its normal approaches. However, the visual investigation of orbital energies and positions can be helpful in for example, deciding whether redox is a likely mechanism or defining positions of reagent attack. Students are given a Frontier Orbital problem to solve, showing why a certain reaction is stereoselective. This MOPAC6 is modified from an old version of Tim Clark's VAMP program - kindly donated before it was commercialised.
Click the button. You are presented with a choice. However, you cannot display something which has not yet been run!. Choose 'R' for run.
Optimisation is usually not necessary as you will have minimised the structure with molecular mechanics already
Enter an output filename, hit enter and wait. MOPAC is carried out on-line so avoid large molecules.
Several files are generated by MOPAC:
.mov is a specialised file which will be used by the display module to be described next
.mpp is the usual full output file from MOPAC6. It is a text file and can be scanned in the editor
.vmp is a XED/COSMIC file which can be read into the main screen.
This MOPAC6 version was prepared by Tim Clark at Erlangen in response to learning about XEDs. It produces Natural Atomic Orbital (NAOs). These equate quite well with XEDs and were originally used to validate XED parameterisation. They have since been discarded on the basis that they are too complex and do not behave well when two or more molecules are interacting (see XEDOCK - Dock 2 Molecules). They are nevertheless of use under certain conditions
The special window in which the MOPAC has been run, can now be killed (top left bar or <alt> -close)
NB. If the .vmp file cannot be read, it will contain information about why it failed to be produced properly.
Click the MOPAC button. You are presented with a choice. Choose 'D' for display.
When you have run a molecule through MOPAC, the necessary files will be available to view the results. Enter the simple data file used to input into MOPAC originally. (Note- asking for an input file rather than using the screen molecule is because you may want to look at a different set from the current screen molecule).
The routine now asks for the filename of the .mov file. As above, it is only necessary to enter the name, not the extension. This is often the same as the data file asked for first.
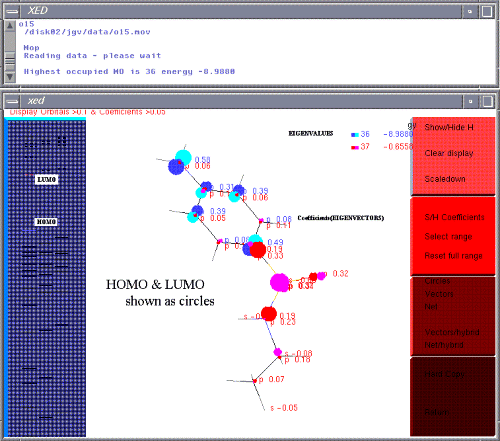
You should be confronted with a new screen featuring eigenvalues (orbital energy levels) on the left and a few open book pages on the right.
Solid eigenvalues are occupied orbitals - HOMO is therefore at the top of the solid lines. Dotted eigenvalues are virtual orbitals - LUMO is therefore at the bottom of the dotted lines.
Click on HOMO. Click on LUMO. You can display up to 16 orbitals but the information is rather overbearing. The energies (eV) are displayed on the top right as each orbital is picked. Orbitals are colour coded up to 24 colours before recycling.
Rotation is as described for the main window but varies in two aspects. A full screen is available during rotation and the cursor must be dragged above the window to return to the books. Below and to the sides does not work.
Book 3 allows different orbital representation. The 'net' display is the most accurate- simulating real Slater-Type Orbitals as far as possible. Hybrid orbitals are best used when there is significant d orbital contribution - otherwise little difference will be seen between unhybridised and hybridised orbitals
Only orbitals are shown with a coefficient greater that 0.1. By toggling 'S/H Coefficients' (S/H = show and hide toggle), coefficients to 0.05 are written against the atoms.
From Book 2, you can select a range of eigenvalues to display. This can be useful for large molecules which have many eigenvalues. By selecting an upper and lower limit, a simpler picture is presented.
