
MacroModel is most conveniently used with the Maestro interface.
- Documentation as PDF files in the directory: /usr/glea/schrodinger/docs/mm8/pdf/
- Long calculations, such as conformation searches and molecular dynamics should always
be run by submitting to the batch queue. When the calculation has been set up, do not click
Start but click on Write Job Files instead. This creates files called
filename.mae and filename.com. Type bminbat filename on the
command line to send the job to the batch queue.
- Helpful commands
- bminbat filename: Submits calculations to the batch queue
- mmck filename: Checks the progress of a conformation search
Once a molecule has been imported to or built in Maestro, MacroModel will do
force-field energy calculations on it. The simplest of these
is a single point calculation
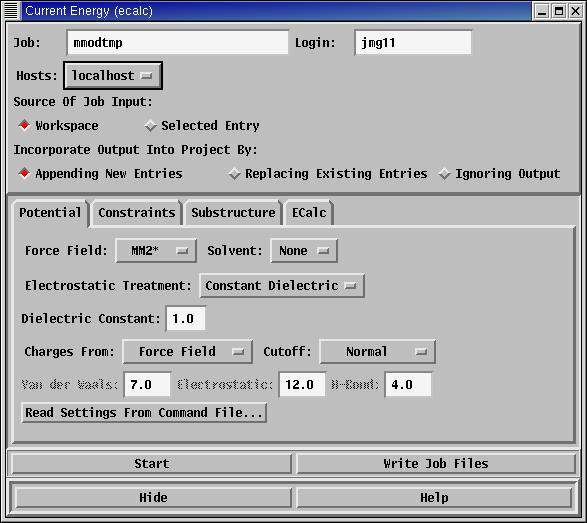
Current Energy: (Menu: MacroModel->Current Energy...)
This will calculate the energy of the molecule, exactly as it is drawn. Selecting
this option from the menu will create a new panel, allowing you to select a
filename for the calculation (default: mmodtmp), a force field
(default MM2*) and to start the calculation. A new window
will appear (Monitori), giving the results of the calculation, including any errors if the
force field does not recognise all of the molecule. The results of the calculation
are also written to files:
mmodtmp.com mmodtmp.log mmodtmp.mae mmodtmp-mon.mae mmodtmp-out.mae
This will overwrite any earlier versions of the files, so it is important to choose
good filenames. This replaces 'ECalc' from earlier versions of MacroModel.
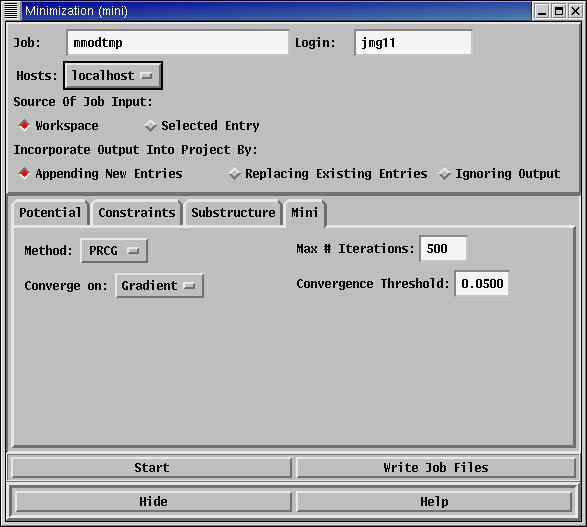
Minimisations: (Menu: MacroModel->Minimization...)
A molecule that has been built in Maestro
will generally not have a perfect geometry, and so the result of
a single point calculation is likely to be a big number which relates
to the precise molecular geometry which has been given to MacroModel.
It would be much more useful to be able to draw an approximate geometry
and have MacroModel turn it into a better geometry. Minimisation is
the process of making small changes to the geometry so that the energy
goes down, and stops when all small changes result in an increase in
energy.
Choosing this option (Menu: MacroModel->Minimization...) gives a similar panel
to that seen for single point calculations, but with rather more options. Pressing the
start button will again generate the Monitori panel, which will follow the calculation.
The geometry of the structure in the workspace will change as the calculation progresses.
The energy that results from minimisation is usually more useful than that from a
single point calculation, as anybody who sketches a simple molecule, for example, ethane,
and minimises it with the same choice of force field and other parameters, should
always get the same answer.
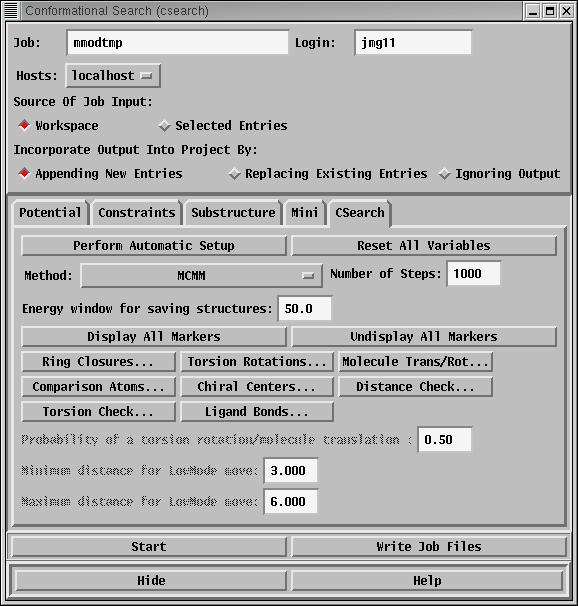
Conformation search: (Menu: MacroModel->Conformational Search...)
Minimisations do not always give the results you might expect,
however. If two people draw cyclohexane and minimise it, one
might get a chair conformation and the other a twist boat. These
have different energies. For most molecules it is necessary not
only to minimise a geometry but also to explore the conformation
space of the system, to ensure that all minima have been found.
The conformation search option does this.
|