Practical Five: Viewing Orbitals and Normal Modes
(1) Orbitals
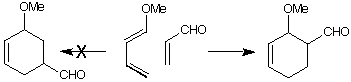
The Diels Alder reaction of methoxybutadiene with acrolein preferentially
gives the ortho adduct. In this practical, molecular orbital calculations will
be used to rationalise this result.
 (a) Build the two reactants as separate molecules using MacroModel and
optimise them using the MM2 force field. MM2 will put lone pairs on the
methoxy oxygen, which are not needed for the molecular orbital
calculation. They must be deleted before the minimised structures are
written to files. Write each structure to a different file. Stop MacroModel
running.
(a) Build the two reactants as separate molecules using MacroModel and
optimise them using the MM2 force field. MM2 will put lone pairs on the
methoxy oxygen, which are not needed for the molecular orbital
calculation. They must be deleted before the minimised structures are
written to files. Write each structure to a different file. Stop MacroModel
running.
(b) Type 'xed' to run
the Xed molecular modelling package.
In this practical, you will use only a small part of this program's
capabilities. The relevant page of the manual is
available. Read in the file.
Xed can read MacroModel files. Move the mouse to the INPUT/OUTPUT area,
and select 'R/W macromodel'. Note that Xed will expect the file to be
in your home directory, and not the current directory.
Once the structure has been read in, move the mouse to the
PROCEDURE/MULTIFILES area. Click on 'Mopac-Run & Look'.
Type 'R' for Run, and the choose the default option (No Optimisation).
Your structure has already been optimised by MacroModel, and this
does not need to happen again. You will be asked for a filename, and
you must choose one that you have not used before. It may be a good
idea to put '_mo' and the end of the filename, to remind you that it
is a molecular orbital calculation.
When the calculation is complete, click on 'Mopac-Run & Look' again,
but this time choose 'D' for display. You will be asked for a filename,
and you must type the name of the original macromodel file, including the
'.dat' You will be asked for a second filename containing the eigenresults.
This will be the name that you used for the MOPAC calculation (probably ending
'_mo'). Type this name, followed by '.mov'. For example, if your MacroModel
structure was called acrolein.dat and the MOPAC calculation filename was
acrolein_mo, then you should type first 'acrolein.dat' and then
'acrolein_mo.mov'
(c) Examine the results. This
will draw your molecule, and let you add the molecular orbitals to it. The
left hand side of the screen shows the energies of the orbitals graphically.
Clicking on an orbital will add it to the molecule displayed in the centre of
the screen. The Highest Occupied Molecular Orbital (HOMO) and the
Lowest Unoccupied Molecular Orbital (LUMO) are the most important
ones. The energies of these orbitals will be displayed when you select
them. Occupied molecular orbitals are represented as full lines, and
unoccupied orbitals as dotted lines.
(d) You can rotate the molecule while the orbitals are displayed. To get
back to the menus, move the mouse into the text window at the top
of the screen. If this does not work, try pressing 'Esc' or the
right hand mouse button. 'Clear display' will remove the orbitals
from the molecule, but continues to display the molecule itself.
The mouse buttons for rotation are slightly different to that for MacroModel. Moving the mouse whilst holding down the left
mouse button makes the molecule rotate. Holding the
right mouse button translates the molecule. The middle button rotates
around an axis coming out of the screen.
(e) Will HOMO of the butadiene react with LUMO of the dienophile- or the
other way round? Look at the energy differences. Which way round will the
molecules prefer to react? (Look for the best orbital overlap.)
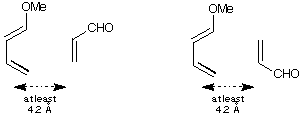
(f) Build the two combined structures (acrolein up and acrolein down) with
the two reactants within roughly the distance shown (not too close!) and
repeat the calculation using MOPAC. Now see where HOMO and LUMO
appear and display the coefficients (orbital sizes). You can move the
molecules relative to each other by clicking on TRMol then
selecting one of the molecules. A small cross will appear, and you can
move this molecule by clicking on the cross with the middle mouse button
and holding the button down. Moving the mouse will move just the
selected molecule. If you hold down the shift key, you will rotate just this
molecule.
 (g) Can you also explain the relative stereochemistry of the product?
(g) Can you also explain the relative stereochemistry of the product?
(2) Normal Modes
A minimum energy conformation of a molecule will not be still, even at
absolute zero of temperature, but will wobble around. These movements
can be separated into a series of simple motions, called the normal
modes, each of which corresponds to a particular frequency. Analysis of
the normal modes allows us to see which motions have low energy, and
which are less accessible.
Some examples are in the directory /usr/local/examples/NMA To see
them, use the program Eadfrith. From your home directory, type 'render
/usr/local/examples/NMA/filename' where filename is one of the files.
Make sure that NMA (option G) is switched on. When the molecule
appears, you can see its normal vibrational modes by moving the mouse
around with no buttons pressed. Pressing the mouse buttons manipulates
the molecules in the normal way.
Normal mode analyses are given for water, methane, and a formaldehyde
H2BCl complex. They were all calculated using ab initio molecular orbital
theory. One example is available on line, if your
browser will display multiframe gif images.
|